PCR(聚合酶链式反应)几乎是分子实验室里最常见的技术之一。但有时候,做着做着,条带就是不上镜:要么无条带,要么全是拖尾,要么阴性对照还神奇地亮了……🤯
别急!今天我们就来盘点 PCR失败的5个常见原因,并提供实用的解决办法,帮你少踩坑。
1️⃣ 模板DNA质量或浓度不合适
🔍 表现:无条带、条带过弱或拖尾。
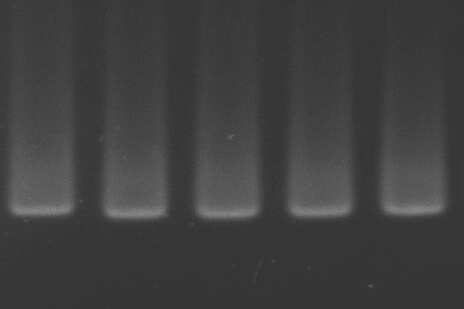
📌 原因:DNA降解、杂质残留,或者浓度过低/过高。
✅ 解决办法:
- 保证DNA纯度(A260/A280 = 1.8–2.0);
- 稀释过浓模板;
- 植物/土壤等样本建议用高纯度提取试剂盒。
2️⃣ 引物设计或浓度有问题
🔍 表现:非特异性条带、引物二聚体。
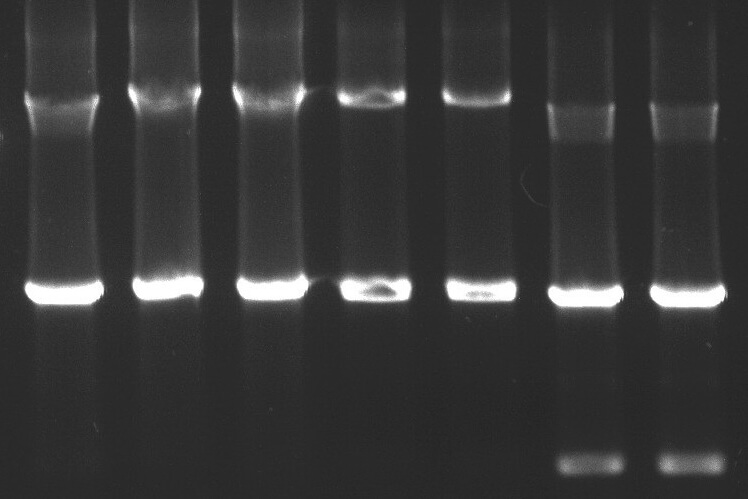
📌 原因:引物设计不合理,浓度过高/过低。
✅ 解决办法:
-
长度18–25 bp,GC含量40–60%;
-
避免3’端互补;
-
终浓度一般0.1–0.5 μM,必要时做梯度PCR优化。
3️⃣ PCR体系或酶活性问题
🔍 表现:完全无扩增或扩增效率低。

📌 原因:Taq酶失活、Mg²⁺浓度不合适、dNTP降解。
✅ 解决办法:
-
分装保存酶,避免反复冻融;
-
优化Mg²⁺浓度(1.5–2.5 mM);
-
用新鲜dNTP;
-
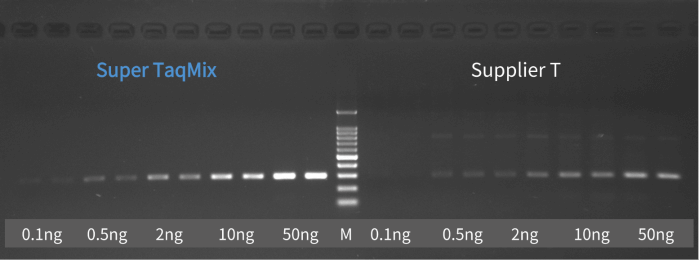
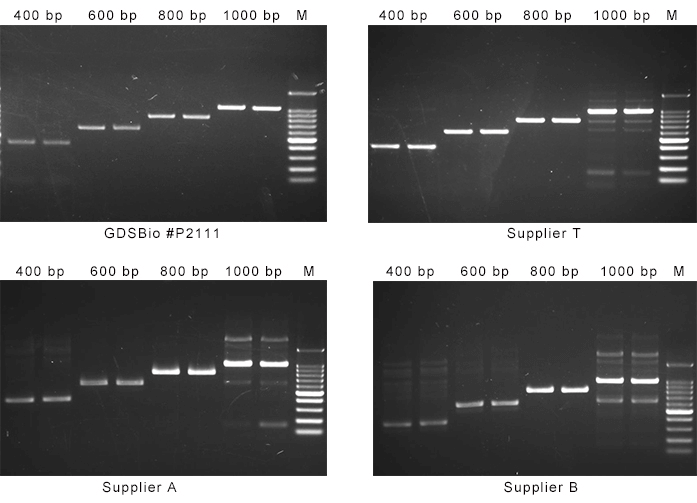
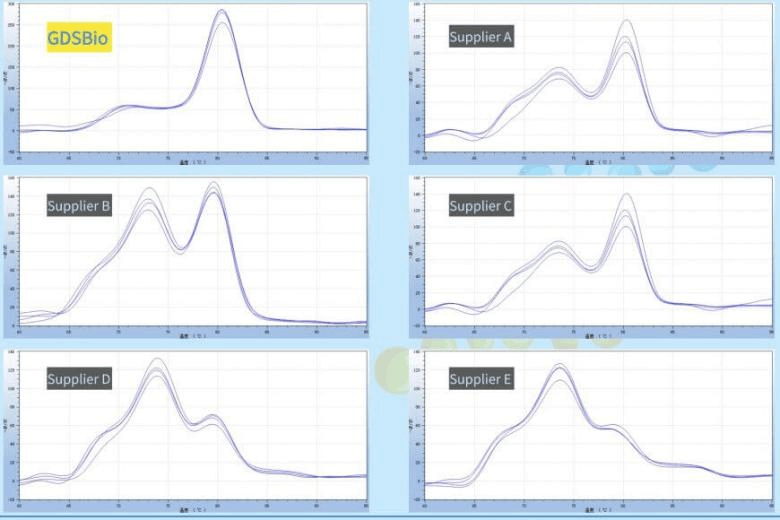
推荐选择性能优异的PCR预混液 👉 预混液中已优化了缓冲体系和酶活性,能显著提升扩增成功率,减少反复调试的时间,非常适合常规科研实验。
4️⃣ 实验操作污染
🔍 表现:阴性对照也出现条带。
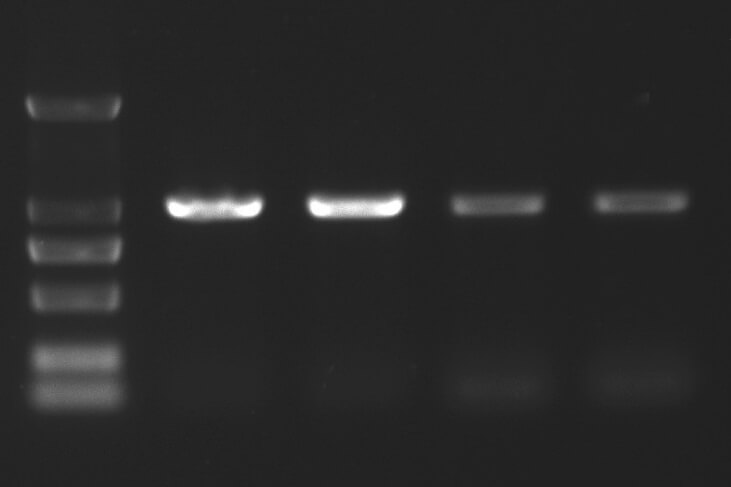
📌 原因:PCR产物残留、移液枪污染、空气中DNA污染。
✅ 解决办法:
-
严格分区操作(提取区/体系配置区/产物分析区分开);
-
用带滤芯的枪头;
-
定期清洁实验台,使用核酸去除剂。
5️⃣ 循环条件设置不当
🔍 表现:条带模糊、非特异性扩增或无结果。
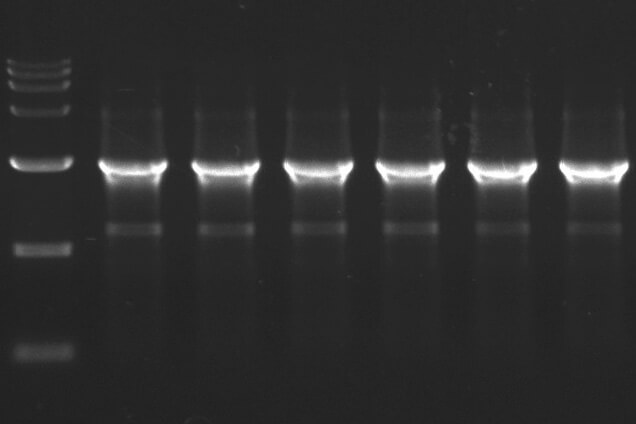
📌 原因:退火温度过低/过高、延伸时间不足、循环数过多。
✅ 解决办法:
-
根据引物Tm值设置退火温度;
-
延伸时间约 1 kb/min;
-
循环数控制在30–35次。
🧪 总结
PCR失败并不可怕,常见问题主要集中在 模板、引物、体系、操作和循环条件 这五个方面。逐一排查,总能找到原因。
而对于高校实验室或科研人员来说,选择一款高性能PCR预混液,往往能让PCR事半功倍,节省大量试错时间。
🔬 科研路上,少一点弯路,多一点高效!
| 货号 | 产品名称 | 适用场景 |
| P2141 | Super TaqMix | 常规PCR扩增 |
| P2111 | Super HIFI PCR Master Mix | 长片段高保真扩增 |
| P2121 |
|
|
| P2091 | SYBR Green qPCR Mix | 染料法高特异性基因定量分析 |
| P2701 | Multiplex Probe qPCR Mix Plus U | 探针法多基因同时定量检测 |
| R1081 | PowerScript RT SuperMix | 全预混高效RNA逆转录 |
| RP1001B | 2X One Step RT-PCR Mix | 快速逆转录与PCR扩增一体化 |
| V5007 | 2X One Step Prime RT-qPCR Mix | 快速灵敏的转录组基因定量检测 |
| P9013 |
|
|
|
|
|
|
| R1021 | TRAzol | 高效提取高质量RNA |
订购咨询
广州东盛生物科技有限公司
📧 sales@gdsbio.com
📞 020-87791356
🌐 www.gdsbio.cn